TABLEAU COMPARATIF
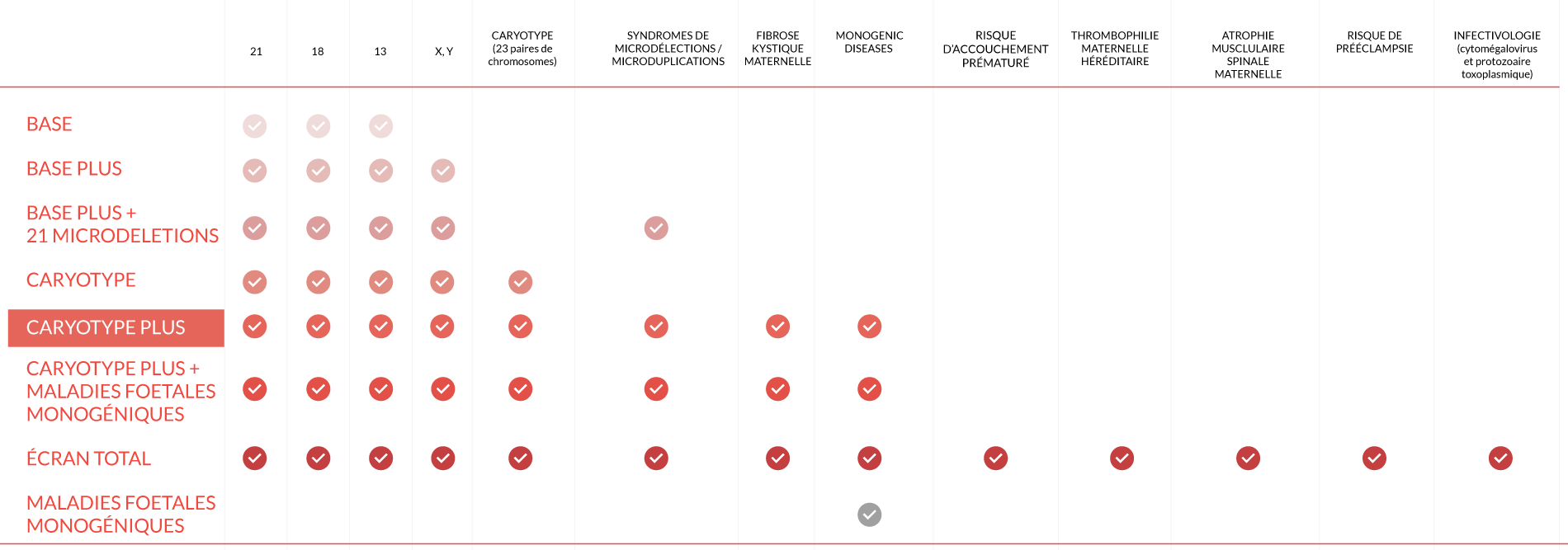
FetalDNA Karyotype Plus est l’un des niveaux les plus complets en tant que test prénatal : il ajoute à toutes les investigations précédentes le screening d’un grand nombre d’altérations chromosomiques causées par des réarrangements structurels (qui sont définis comme des microduplications / microdélétions) à une résolution d’environ 7 Mb (pour les résolutions, un diagnostic inférieur sur le sang maternel n’est pas obtenu, seul un diagnostic prénatal invasif tel que l’amniocentèse ou le CVS doit être utilisé, en réalisant une étude spécifique avec des puces à ADN).
En bref, avec le niveau Karyotype Plus, les altérations numériques des 23 paires de chromosomes du fœtus sont analysées (y compris les trisomies des chromosomes 13, 18, 21 et les anomalies des chromosomes sexuels X et Y, déterminant également le sexe fœtal sur demande) et, grâce à une évaluation bioinformatique particulière, il est également possible d’inspecter la structure interne des chromosomes, avec définition de l’ordre des mégabases.
FetalDNA Karyotype Plus est également en mesure d’élargir l’investigation des pathologies avec un screening qui permet d’obtenir des informations sur la présence des syndromes de microdélétion les plus importants chez le fœtus.
Le terme microdélétion / microduplications fait référence à des anomalies caractérisées par l’absence d’un petit tractus chromosomique entraînant une perte d’informations génétiques (microdélétions) ou par l’ajout de matériel génomique surnuméraire (microduplications). Les deux conditions provoquent des pathologies avec des images cliniques et phénotypiques complexes et variables selon le chromosome impliqué, la région chromosomique impliquée et la taille de la microdélétion / microduplication elle-même.
Les principaux syndromes de microdélétion étudiés par le FetalDNA Karyotype Plus sont listés ci-dessous:
Ainsi, si l’une de ces mutations est présente chez la mère, il faudra rechercher si le fœtus est également porteur simple ou, si le père était également porteur, a couru le risque d’être atteint de fibrose kystique. Cela se produit dans 25 % des cas si les deux parents étaient porteurs sains.
Avec le FetalDNA Karyotype Plus, l’analyse du gène maternel est réalisée à travers un dépistage appelé 1er niveau qui permet d’analyser les mutations les plus courantes et les plus fréquentes, parvenant à identifier environ 83% des porteurs. La fréquence estimée, dans la population italienne, des porteurs sains (souvent inconscients de l’être) est de 1 sur 25-30, celle des naissances atteintes est de 1 sur 2500 – 3000.
Sexe de l’enfant disponible sur demande.