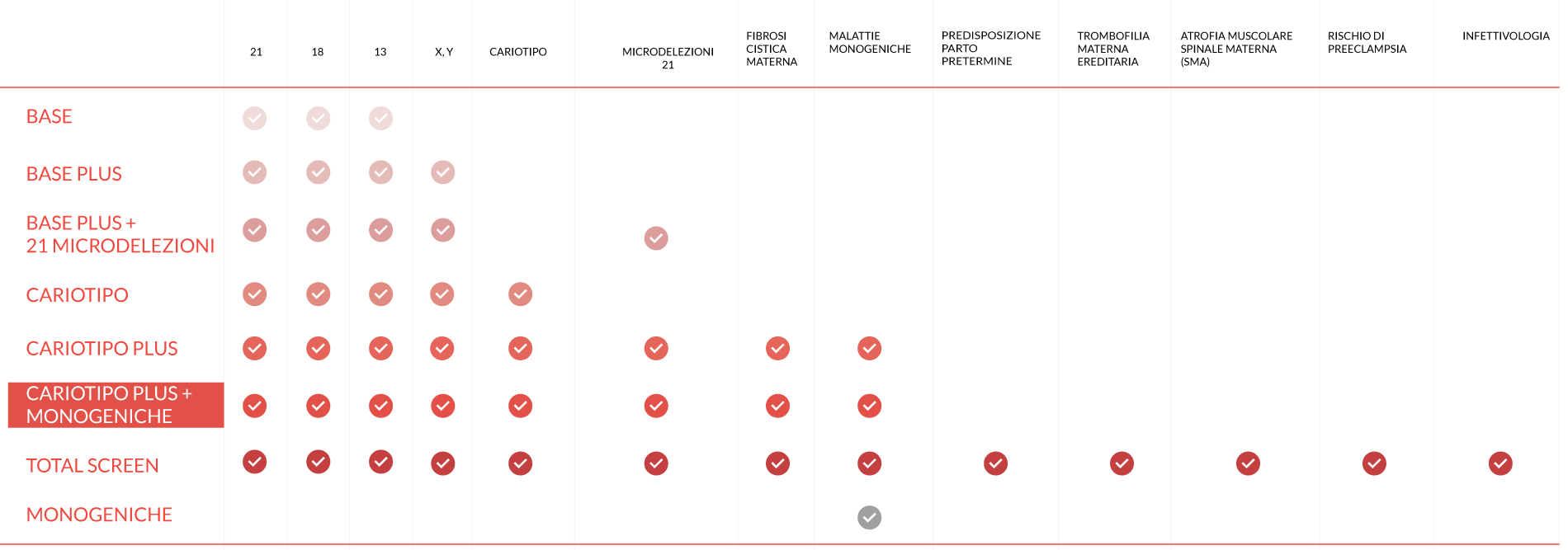
TABELLA COMPARATIVA
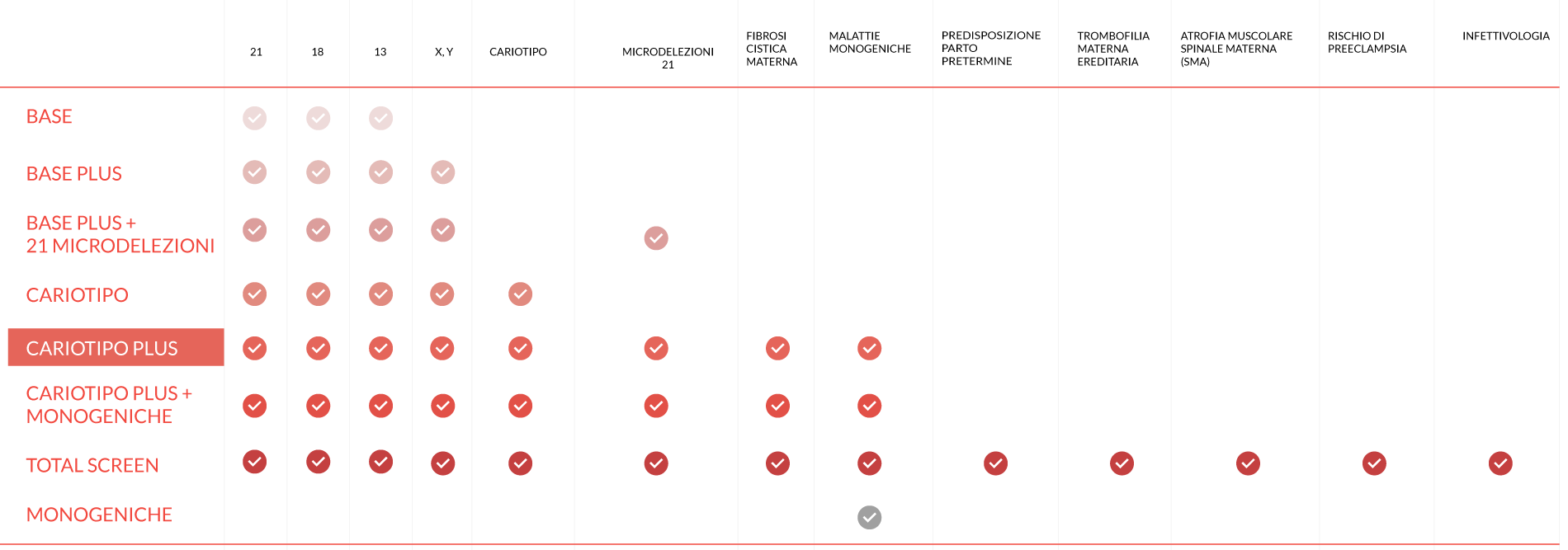
FetalDNA Cariotipo Plus è uno dei livelli più completi come test prenatale: aggiunge a tutte le indagini precedenti lo screening di un gran numero di alterazioni cromosomiche determinate da riarrangiamenti strutturali (che vengono definite microduplicazioni / microdelezioni) ad una risoluzione di circa 7 Mb (per risoluzioni inferiori non si ottiene diagnosi sul sangue materno si deve ricorrere solo alla diagnosi prenatale invasiva, Amnio o Villocentesi, eseguendo uno studio specifico con microarrays).
In sintesi, con il livello Cariotipo Plus si analizzano le alterazioni numeriche delle 23 coppie di cromosomi del feto (incluse le trisomie dei cromosomi 13, 18, 21 e le anomalie dei cromosomi sessuali X e Y, determinando, anche il sesso fetale su richiesta) e, grazie ad una particolare valutazione bioinformatica, si può ispezionare anche la struttura interna dei cromosomi, con definizione dell’ordine delle megabasi.
FetalDNA Cariotipo Plus riesce inoltre ad ampliare l’indagine sulle patologie con uno screening che consente di ottenere informazioni sulla presenza nel feto delle più importanti sindromi da microdelezione.
Il termine microdelezione/microduplicazioni si riferisce ad anomalie caratterizzate dall’assenza di un tratto cromosomico di piccole dimensioni con conseguente perdita di informazione genica (microdelezioni) o dall’aggiunta di materiale genomico sovrannumerario (microduplicazioni). Entrambe le condizioni causano patologie con quadri clinici e fenotipici complessi e variabili dipendenti dal cromosoma coinvolto, dalla regione cromosomica interessata e dalle dimensioni della microdelezione / microduplicazione stessa.
Di seguito vengono elencate le principali sindromi da microdelezione indagate dal FetalDNA Cariotipo Plus:
In tal modo, se fosse presente una di queste mutazioni nella madre, si dovrà indagare se il feto sia anch’egli un semplice portatore o, qualora fosse portatore anche il padre, corresse il rischio di essere affetto dalla Fibrosi Cistica. Ciò avviene nel 25% dei casi se tutti e due i genitori fossero portatori sani.
Con il FetalDNA Cariotipo Plus viene effettuata l’analisi del gene materno attraverso uno screening chiamato di 1° livello che permette di analizzare le mutazioni più comuni e frequenti riuscendo ad identificare circa l’83% dei portatori. La frequenza stimata, nella popolazione italiana, dei portatori sani (spesso inconsapevoli di esserlo) è di 1 su 25–30,quella dei nati affetti è di 1 su 2500 – 3000.
Su richiesta può essere fornito anche il sesso fetale.